 2023, Vol. 41
2023, Vol. 41Institute of Oceanology, Chinese Academy of Sciences
Article Information
- WANG Xinran, WANG Junhao, ZHU Yunke, ZHU Xinyu, QIN Hong, BIAN Ka, TANG Xianghai
- Comparison in structure and predicted function of epiphytic bacteria on Neopyropia yezoensis and Neopyropia katadae
- Journal of Oceanology and Limnology, 41(6): 2232-2248
- http://dx.doi.org/10.1007/s00343-023-3078-5
Article History
- Received Apr. 30, 2023
- accepted in principle May 18, 2023
- accepted for publication Jun. 30, 2023




Neopyropia is a red alga that has a long history of cultivation in Asia. It is one of the most important cultivated seaweeds in China because of its unique nutritional value and edible flavor (Yoshida et al., 1997). The thalli of Neopyropia yezoensis mostly grow on intertidal rocks, and with the tide, they are exposed to the air and forced to desiccate when the tide recedes, and the water loss rate is as high as 90%. However, at high tide, the blade is resubmerged in seawater and grows normally (Davison and Pearson, 1996), with a strong ability to tolerate dehydration and rehydration. Neopyropia katadae is often found in puddles among rocks in the intertidal zone and remains underwater even at low tide. Its tolerance to desiccation is much lower than that of N. yezoensis, and the threshold for tolerance to water-loss stress is 42%–46% (Lin et al., 2009; Wang et al., 2016). This difference in water loss tolerance may be due to the differences in the molecular mechanisms of stress resistance among algae. It is generally thought that nori under daily dry exposure after re-entering the water surface can eliminate the attachment of miscellaneous algae, floating mud, and some bacteria, thus promoting the healthy growth of algae, which is a natural phenomenon beneficial to algae. However, there is no such periodic water loss in the life history of N. katadae, and the loss of water could kill the algae. Theoretically, N. yezoensis can also grow in water for a long time. However, in fact, the health status of N. yezoensis in this case is poor.
Algae and bacteria have coexisted in the ocean for more than 200 million years (Falkowski et al., 2004) and are closely related. Marine microorganisms are affected by many environmental factors such as changes in species distribution, habitat degradation, high disease incidence, and species extinction, which have a huge impact on the abundance, distribution, and function of marine planktonic microorganisms (Bulleri and Chapman, 2010; Cook et al., 2011; Campbell et al., 2015; Egan and Thomas, 2015). Macroalgae attract and promote beneficial bacterial species to settle and colonize their surfaces. These epiphytic bacteria help algae combat invasion and colonization by harmful microorganisms by secreting secondary metabolites and/or antimicrobial agents (Goecke et al., 2010). Many studies have shown that the bacterial community composition varies with algal species and physiological status (Schäfer et al., 2002). Lingulodinium polyedra is a B1 and B12 auxotroph. Non-axenic cultures of L. polyedrum can acquire sufficient quantities of vitamins from the associated bacterial community to sustain the maximum growth rate defined by the culture conditions. The growth of the associated heterotrophic bacterial community is sustained by substrates provided by L. polyedrum (Cruz-López and Maske, 2016). Environmental factors also affect the selection of algae for bacteria. When a thallus undergoes desiccation, its surface water gradually evaporates, forming a water film. With the extension of the desiccation time, the water film will gradually become thinner and disappears. During this period, the temperature, pH, and salinity of the water film change (Mercado and Niell, 2000; Ji et al., 2016). These changes in physicochemical properties inevitably affect the combination of bacteria with the algal body, thus impacting the community structure of microorganisms on the surface of the alga.
To study the differences in the structure and abundance of surface bacteria of two species of Neopyropia at the same tidal level and the possible reasons for these differences, and to avoid the errors caused by measuring at a single location, the study used 16S rRNA gene sequence analysis and real-time quantitative PCR to analyze the samples collected from two sea areas in Qingdao (China) and to document the surface bacterial community structure of N. yezoensis with desiccation phenomenon, comparing it with the surface bacterial community of N. katadae without desiccation to explore the mutual selection of different species of bacteria and algae. This study lays the foundation for resolving the close association between surface microorganisms and algal bodies and the physiological and ecological characteristics of nori species.
2 MATERIAL AND METHOD 2.1 Study site and sample collectionNeopyropia yezoensis and N. katadae thalli and seawater samples were collected from two coastal areas in Qingdao, Shandong Province, China. The first location was Qingdao Polar Ocean World (36.06°N, 120.44°E), and the second location was the Haitian Center (36.05°N, 120.36°E). On March 1, 2022, at a water temperature of 5.1 ℃, N. katadae thalli and N. yezoensis thalli were collected from Qingdao Polar Ocean World at the same tide level. The thalli were rinsed in seawater to remove attached sand, placed in sampling bags containing a small amount of local seawater, and transported to the laboratory in ice boxes. In addition, approximately 5 L of seawater was collected and transported. On March 5, 2022, using the same collection method, algae, and seawater samples at water temperature of 5.3 ℃ were collected from the Haitian Center in Qingdao, Shandong Province, China.
2.2 Material processing and groupingTo analyze the microbial communities, thalli (n=8) and seawater (n=4) samples were collected from the two locations. Suitable samples selected from the algae were washed in sterilized seawater to remove the adhering sediment from the surface. Next, DNA was obtained by DNA extraction performed on the surface microorganisms of the algae. The DNA samples were put into 1.5-mL centrifuge tubes and stored at -80 ℃. In addition, debris was collected from each 600-mL seawater sample by repeated filtration through a nylon mesh (200-µm pore size) and then through a 0.22-µm polycarbonate film to obtain the microbial biomass, and no less than 500 mL of water samples were filtered. Four filter membranes obtained from each location and the membranes were folded and placed in lyophilization tubes for storage at -80 ℃.
Water, N. katadae, and N. yezoensis algal samples collected from Qingdao Polar Ocean World were designated as groups M1, B1, and T1, respectively; and those obtained from the second location, the Haitian Center, were designated as groups M2, B2, and T2, respectively.
2.3 DNA extraction and subsequent processingFor the extraction of bacterial community DNA from the thalli and seawater samples, a Precellys® 24 (Bertin Corp., Rockville, MD, USA) and DNeasy® PowerSoil® kit (Qiagen, Hilden, Germany) were used. The DNA was stored at -20 ℃ until amplification. The DNA concentration and purity were monitored on 1% agarose gels, and all DNA samples were diluted with sterile water to adjust the concentrations to 1 ng/µL (Ahmed et al., 2021).
For amplification, the V3-V4 hypervariable region of the 16S rRNA gene was targeted using primer pairs 341F (5′-CCTAYGGGRBGCASCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAA-3′) (Wagner et al., 2014). The reactions were performed using Phusion® High-Fidelity PCR Master Mix (New England Biolabs) and efficient high-fidelity enzymes for PCR. PCRs were conducted in a final reaction volume of 50 μL containing 15 μL of 2× Phusion® Master Mix, 1-μL each of forward and reverse primers, 10 μL of gDNA (1 ng/μL), and 3-μL ddH2O. The initial denaturation was performed at 98 ℃ for 1 min, followed by 30 cycles of denaturation at 98 ℃ for 10 s, annealing at -50 ℃ for 30 s, and elongation at 72 ℃ for 30 s, and then finally 72 ℃ for 5 min. Each DNA sample was amplified three times. The PCR products were mixed in equal amounts according to the concentration of the PCR products and then purified by 2% agarose gel electrophoresis. The target bands were recovered using a Universal DNA Purification Recovery Kit.
The library was constructed using a NEBNext® UltraTM DNA Library Prep Kit (New England Biolabs) and tested and quantified by qPCR using an Agilent 5400 device. A NovaSeq 6000 system was used for sequencing.
2.4 Data processing based on bioinformatics and statistical analysisRaw data obtained from sequencing were spliced and filtered to obtain clean data. Then, noise reduction was performed using Divisive Amplicon Denoising Algorithm 2 (DADA2), and sequences with an abundance of less than five were filtered out to obtain the final Amplicon Sequence Variants (ASVs) (Callahan et al., 2016). On the one hand, species annotation was performed for each ASVs sequence. The ASVs were analyzed for abundance, alpha diversity, and Venn diagrams. Subsequently, differences in the community structure between samples or groups were explored using methods of reduction dimensions, such as PCA and NMDS. To further explore the differences in community structure among grouped samples, statistical analysis methods such as the t-test, MetaStat, and LEfSe (LDA Effect Size) were selected to test the significance of differences in species composition and community structure of grouped samples. The annotation results of amplicons can also be correlated with the corresponding functional databases, and the PICRUSt2 software can be selected for functional prediction analysis of microbial communities in ecological samples.
The sample data were separated from the downstream data based on barcode sequences and PCR amplification primer sequences, and the reads were spliced using FLASH (V1.2.11; http://ccb.jhu.edu/software/FLASH/) (Magoč and Salzberg, 2011) to obtain raw tags after truncating the barcode and primer sequences. The obtained Raw Tags were then quality-controlled using fastp software (V0.20.0). Finally, the Clean Tags were compared against database using the Usearch software to detect chimeras and remove them (Haas et al., 2011) to obtain the final validated data, i.e., Effective Tags. For the above obtained Effective Tags, the DADA2 module or deblur in QIIME2 software was used for noise reduction (DADA2 was used by default), and sequences in abundance of less than five were filtered out to obtain the final ASVs as well as the feature tables. Subsequently, the obtained ASVs were compared with the database using the classify-sklearn module in the QIIME2 software to obtain the species information of each ASV using Silva database 138.1 annotations (Bolyen et al., 2019).
Alpha diversity was used to analyze the microbial community diversity within the sample community (Li et al., 2013). Alpha diversity analysis, conducted on individual samples, offers valuable insights into the richness and diversity of microbial communities within each sample. It encompasses the assessment of species richness and diversity variations among samples through the utilization of diverse statistical analysis indices, species diversity curves, and species accumulation boxplots. These analytical tools collectively aid in evaluating the differences in species abundance and diversity within microbial communities across various samples.
Beta diversity represents a comparative analysis of the microbial community composition of different samples. First, UniFrac distances were calculated using QIIME2 software, and R software was used to plot the PCA, PCoA, and NMDS downscaling. Among these, PCA, PCoA, and NMDS are called the ade4, ggplot2, and vegan packages in R software. Subsequently, the adonis and anosim functions in QIIME2 software were used to analyze the significance of community structure differences between groups. Finally, the analysis of significantly different species between groups was performed using LEfSe and R software. MetaStat analysis was performed using R software to obtain P values for the two comparison groups at six taxonomic levels (phylum, class, order, family, genus, and species), and the species with P values less than 0.05 were selected as significantly different species between groups. The t-test was also used in the R software to analyze significant differences in species at each taxonomic level.
PICRUSt2 is a computational tool that can use 16S amplicon data to predict the functional profiles of microbial communities (Langille et al., 2013; Douglas et al., 2019). The analysis by the PICRUSt2 (V2.3.0) method is based on the gene information regarding the ASV tree and ASVs in the Greengenes database (V1.9.1) to infer the gene function profile of their common ancestor and to infer the gene function profile of other unknown species in the Greengenes database, constructing the gene function prediction profile of the whole spectrum of archaea and bacterial domains, and finally "mapping" the composition of the sequenced bacterial groups to the database to predict the metabolic function of the bacterial groups. This is possible because certain functional traits are often conserved across related microbial taxa. By referencing known functional annotations from reference genomes, PICRUSt2 maps the 16S rRNA gene sequences to a phylogenetic tree and assigns functional annotations based on related taxa. It extrapolates functional information from reference genomes to closely related taxa, providing insights into the potential functional capabilities of microbial communities based on their taxonomic composition.
2.5 Real-time quantitative PCR detection systemTo investigate the relative relationship between the number of bacteria on the surface of N. yezoensis thalli and N. katadae thalli, the thalli samples were subject to qPCR assay, and the weight of the algae was controlled to be equal when extracting DNA, both being 0.8 g.
As the percentage of bacteria in the genus Granulosicoccus in each sample is known, we chose the specific sequence of this bacterium to design primers, calculated the copy number of this genus by absolute quantification, and then inverted the number of all bacteria on the blade by the percentage.
Plasmids containing correctly sequenced target genes were selected, and plasmid concentrations were measured using a NanoDropTM device. According to the formula: copies=6.02×1023 (copies/mol) ×plasmid concentration (ng/μL)×10-9/[(molecular weight of the inserted fragment+molecular weight of T vector)× 660] (g/mol), and the concentration of recombinant vector plasmid was then converted and diluted with double-distilled water, totaling five concentrations.
The primer sequences for the qPCR were Gra-F: (5′-TGCAAGCGTTAATCGGAATTACTGG-3′), Gra-R: (5′-TCTACGCATTTCACCGCTACACC-3′). Reaction system: 2× ChamQ SYBRTM Color qPCR Master Mix (High ROX Premixed) 10 μL, forward and reverse primers 0.4 μL each, DNA 1 μL, and ddH2O 8.2 μL. Reaction procedure: 95 ℃ for 30 s; 40 cycles of 95 ℃ for 10 s, 60 ℃ for 30 s; 95 ℃ for 15 s, 60 ℃ for 60 s, and 95 ℃ for 15 s.
3 RESULT 3.1 Diversity and composition of bacterial communityBased on the species annotation results, 2 217 ASVs were identified at location 1. Among them, 49 ASVs were shared by all three groups, 1 208 ASVs were specific to group M1, 185 ASVs were specific to group B1, and 303 ASVs were specific to group T1. At location 2, 2 150 ASVs were obtained, with 33 ASVs common to all three groups, 1 118 ASVs specific to group M2, and 140 and 373 ASVs specific to groups B2 and T2, respectively (Fig. 1).

|
| Fig.1 The Venn diagram of the three experimental groups a. location 1; b. location 2. |
The bacterial community structure of the samples at the phylum level is shown in Fig. 2a. The dominant phyla in the microbial communities of the two species differed. Proteobacteria was the predominant phylum in the microbial community of N. katadae, accounting for 86.72% and 78.69% of the relative abundance. In contrast, Bacteroidetes was the most abundant phylum in the microbial community of N. yezoensis, accounting for 65.43% and 55.74% of the relative abundance, followed by Proteobacteria with 27.00% and 26.00% of the relative abundance, respectively. A significant difference in the dominant phyla between the microbial communities of the two species was observed through a statistically significant comparison (P < 0.05) (Fig. 2b–c).

|
| Fig.2 Characterization of bacterial community members at the phylum level a. relative abundance (%) of bacterial taxa at the phylum level; b. t-test of differential bacterial populations at the phylum level between B1 and T1; c. t-test of differential bacterial populations at the phylum level between B2 and T2. |
At the genus level, Granulosicoccus was the dominant genus in the microbial community of N. katadae, accounting for 83.47% and 21.17% of the relative abundances at the Qingdao Polar Ocean World and Haitian Center locations, respectively (Fig. 3a). In contrast, N. yezoensis exhibited different dominant genera at these two locations: Olleya (25.53%) and Lewinella (24.37%), respectively. This variation in the dominant genera of N. yezoensis may be attributed to differential exposure to air and the contrasting environmental conditions at the two locations. Furthermore, this observation suggests that the N. katadae microbial community may have greater stability, possibly because of its continuous aquatic habitat, compared to N. yezoensis, which experiences more pronounced changes in response to varying environmental conditions (Fig. 3). A statistically significant difference was observed in the dominant genera between the microbial communities of the two species (P < 0.05). The relative abundances of Tenacibaculum and Lewinella were higher in N. yezoensis than in N. katadae, whereas those of Lewinella, Olleya, Pseudoalteromonas, and Leucothrix were higher in N. yezoensis than in N. katadae (Fig. 3b–c).

|
| Fig.3 Characterization of bacterial community members at the genus level a. relative abundance (%) of bacterial taxa at the genus level; b. t-test of differential bacterial populations at the genus level between B1 and T1; c. t-test of differential bacterial populations at the genus level between B2 and T2. |
LDA Effect Size (LEfSe) is a powerful analytical tool used for the identification and interpretation of high-dimensional biomarkers, such as genes, pathways, and taxa, to compare and assess statistically significant and biologically relevant differences between two or more subgroups (Biomarker) (Segata et al., 2011). Figure 4a presents information on the differential abundance of bacteria at the phylum, class, order, family, and genus levels.

|
| Fig.4 Evolutionary branch diagram and differential bacterial communities at each level a. evolutionary branch diagram of differential bacterial communities or species; b. the LDA value (influence value of linear discriminant analysis) distribution histogram shows bacterial communities or species (Biomarker) with LDA score greater than four. |
A total of 82 biomarkers with an LDA score > 4 was identified. Approximately 50% of the identified biomarkers were found in seawater samples from the two different locations, indicating a diverse and rich microbial community. In the microbial community of N. yezoensis, the phylum Bacteroidetes exhibited the highest LDA score, whereas in the microbial community of N. katadae, the phyla Actinobacteria and Proteobacteria had the highest LDA scores, which is consistent with their respective dominant phyla. This suggests that these microbial taxa contribute significantly to the observed differences between the groups in terms of microbial community composition and diversity (Fig. 4b).
3.3 Analysis of microbial diversityAlpha diversity is commonly used to assess the diversity of microbial communities and indices such as Shannon, Simpson, and Chao1 are frequently employed. Chao1 measures species richness, the Shannon index quantifies species diversity, and the Simpson index characterizes species distribution diversity and evenness within a community (Li et al., 2013). Consistent with the LEfSe analysis, the alpha diversity results revealed that the microbial communities in seawater exhibited the highest diversity (Fig. 5). Overall, the alpha diversity of the N. yezoensis microbial community was higher than that of the N. katadae microbial community (P < 0.05), indicating that the former harbored more diverse and evenly distributed microbial species, which is consistent with the species annotation results. The complex microbial community of N. yezoensis may be associated with the unique lifestyle of its host. Microbial communities with high diversity are highly resilient in fluctuating environments.

|
| Fig.5 Comparisons of alpha diversity indices a. Chao1; b. Shannon; c. Simpson. For Group names please refer to Section 2.2 Material processing and grouping. *: P < 0.05; **: P < 0.01. |
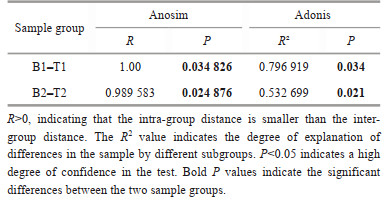
Based on the first two principal components, the microbiota samples of the two species were well-separated, explaining approximately 23% of the total variation (Fig. 6). Compared to N. katadae, the microbiota of N. yezoensis exhibited a more scattered sample distribution, implying a more complex environmental variability experienced by N. yezoensis. ADONIS (Stat et al., 2013) and Anosim (Chapman and Underwood, 1999) analyses revealed that the R-values of the microbiota between the two species were significantly non-zero (P < 0.05), indicating significant differences in the microbiota compositions between the two species (Table 1).

|
| Fig.6 Principal component analysis of samples |
|
Numerous studies have demonstrated the predictive capability of PICRUSt2 for estimating the relative abundance and potential expression of functional gene families in microbial communities using 16S rRNA gene sequencing data (Hartman et al., 2017; Lebrun and Kang, 2018; Ribeiro et al., 2018; Chen et al., 2020).
The abundance of K02014 TC. FEV. OM (iron complex outer membrane receptor protein), K02342 dnaQ (DNA polymerase Ⅲ subunit epsilon), K03924 moxR (MoxR-like ATPase), K03088 rpoE (RNA polymerase sigma-70 factor, ECF subfamily), K06147 ABCB-BAC (ATP-binding cassette, sub-family E, member 1), K02003 ABC. CD. A (putative ABC transport system ATP-binding protein), K02004 ABC. CD. P (putative ABC transport system permease protein), K01990 ABC-2. A (ABC-2 type transport system ATP-binding protein), K01992 ABC-2. P (ABC-2 type transport system permease protein), and K06180 rluD (23S rRNA pseudouridine1911/1915/1917 synthase) were more abundant in the surface microbial community of N. yezoensis (P < 0.05). In contrast, K01091 gph (phosphoglycolate phosphatase), K03406 mcp (methyl-accepting chemotaxis protein), and K00626 ACAT (acetyl-CoA C-acetyltransferase) were more abundant in the surface microbial community of N. katadae (Fig. 7; Supplementary Table S1).

|
| Fig.7 Heat map and t-test results of KO enrichment a. cluster heat map display of KOs based on the predicted gene copy (log10 transformed) of bacterial microbiota across all sample groups; b. t-test of location 1; c. t-test of location 2. |
The pathway map (Fig. 8a) revealed that the microbial samples associated with algal surfaces were enriched for 35 pathways. These pathways included those related to cell composition, such as PWY0-1586 (peptidoglycan maturation) and PWY-5989 (serine biosynthesis Ⅱ), as well as pathways related to energy regulation, such as P105-PWY TCA cycle Ⅳ (2-oxoglutarate decarboxylase) and PWY-5103 (L-isoleucine biosynthesis Ⅲ).

|
| Fig.8 The heat map and t-test results of pathway enrichment a. cluster heat map display of pathways based on the predicted gene copy (log10 transformed) of bacterial microbiota across all sample groups; b. t-test of location 1; c. t-test of location 2. |
Contrary to the predicted KO abundance, the pathway prediction results showed that the abundances of most metabolic pathways were higher in the N. katadae microbial community (P < 0.05). Results from the t-tests conducted at the two locations (Fig. 8b–c) showed that PWY-3781 (aerobic respiration Ⅰ (cytochrome c)), PWY-5667 (CDP-diacylglycerol biosynthesis Ⅰ), and PWY-7664 (oleate biosynthesis Ⅳ (anaerobic)) were more abundant in the microbial community of N. katadae. Only the dTDPRHAMSYN-PWY (dTDP- β -L-rhamnose biosynthesis) pathway was more abundant in the microbial communities of N. yezoensis at both study sites.
3.5 Standard curve and calculation of copy numberBased on the results of pre-amplification of all gradients, standard dilutions of 10-6–10-2, five gradients with the best linear relationship were selected for real-time fluorescent quantitative PCR amplification under optimal reaction conditions to obtain the standard curve and linear regression equation of the amplification reaction. The standard curve showed a good linear relationship between the standard concentration in the range of 10-6–10-2 and the Ct value (the number of cycles experienced in each reaction tube when the fluorescence signal reaches the set threshold value), and the amplification efficiency of the real-time fluorescent quantitative PCR was calculated to be 96.6%, which means that 96.6% of the template was amplified at the end point of each cycle, and the standard curve is shown in Supplementary Fig.S1.
Copies (copies/ng) were calculated based on the Ct values obtained from the standard curve of the samples. As 0.8 g of algae was collected from each sample, the total number of bacteria contained in each sample was deduced based on the percentage of Granulosicoccus in each sample (Fig. 9).

|
| Fig.9 The results of copies calculation for each sample a. the result of location 1; b. the result of location 2. |
Neopyropia katadae and N. yezoensis are two types of seaweed that grow in the same intertidal zone. However, they exhibit distinct differences in their lifestyles: N. katadae stays submerged underwater while N. yezoensis exposes occasionally to the air in tidal zone (Davison and Pearson, 1996; Lin et al., 2009; Wang et al., 2016). Despite their coexistence in the same ecological environment, there is a lack of research on the differences in the microbiota associated with these two seaweed species. The significance of this study lies in filling this knowledge gap, as the comparison of the microbiota between N. katadae and N. yezoensis provides insights into the role of the associated microbiota in the adaptation and survival of seaweeds, which sheds light on the complex interactions between microorganisms and their hosts, and provides fundamental data on the diversity, composition, and function of the microbiota associated with these seaweeds for future research.
4.1 Difference in microbial community structure among groupsThe results revealed differences in the structure and abundance percentages of the microbial communities among the sample groups. A total of 2 217 ASVs were obtained from location 1, and 2 150 ASVs from location 2 (Fig. 1a–b). The diversity of seawater microbiota was higher than that of the epiphytic microbiota in seaweed thalli, as reported in previous studies (Ahmed et al., 2021). The dominant bacterial taxa in the epiphytic microbiota of the two seaweed species differed significantly (Fig. 4). Furthermore, the epiphytic microbiota of N. yezoensis exhibited a higher diversity than that of N. katadae (Fig. 5), which may be associated with the unique living habits of the host (Bulleri and Chapman, 2010; Goecke et al., 2010; Cook et al., 2011) The phyla Proteobacteria and Bacteroidetes were the prominent members of the seaweed microbiota of both species. They are commonly found in aquatic environments (Cottrell and Kirchman, 2000a, b; Biegala et al., 2002; Simonato et al., 2010) and are known to be important associates of algae in general (Tujula et al., 2010).
Bacteroidetes is a major bacterial lineage in marine systems and are among the most abundant microbes in coastal marine waters. It is usually one of the dominant clades in phytoplankton blooms (Alonso et al., 2007). Bacteroidetes, along with its family Flavobacteriaceae, comprised a large proportion of microorganisms in N. yezoensis (Fig. 4a). They are versatile in their ability to degrade a wide range of biopolymers, e.g., being used as a source of carbon and energy (Thomas et al., 2011). Furthermore, Flavobacteriaceae can decompose gelatin and casein, and several species can hydrolyze various polysaccharides, including starch, chitin, pectin, and carboxymethylcellulose (Bernardet et al., 1996). Olleya, a marine bacterium belonging to the family Flavobacteriaceae that produces exopolysaccharides, was more abundant in N. yezoensis than in N. katadae (Nichols et al., 2005). These metabolites of microorganisms may also play an important role in the growth and development of algae (Cosgrove, 2005; Delattre et al., 2011). However, some pathogenic bacteria of the phylum Bacteroidetes can cause harm to algae, and several diseases of macroalgal species have been attributed to members of Bacteroidetes, such as "Anaaki disease", "ice-ice disease", "spot-rotting disease", and "shot hole disease" (Johansen et al., 1999; Thomas et al., 2011). Some pathogenic bacteria use capsules to closely associate with the algal body, making it difficult to separate the bacteria from the algae because of resistance to phagocytosis, drought, and adhesion. Therefore, periodic water loss may play an important role in keeping the healthy growth of N. yezoensis by effectively removing harmful bacteria from the surface of the algae during drying and rehydration. It was also evident that N. yezoensis with frequent dry exposure tended to grow healthier, whereas N. yezoensis without dry exposure was more susceptible to rot. Therefore, the configuration and composition of surface-associated microorganisms may influence the ecological distribution patterns of these two seaweed species.
The Proteobacteria phylum has been found to be more abundant on the surface of N. katadae compared to N. yezoensis in both locations. Proteobacteria are known to have a worldwide distribution in coastal waters (Venter et al., 2004; Rusch et al., 2007) and have been shown to have a possible symbiotic relationship with microalgae, as they can produce and provide vitamins B1 and B12 to algae in exchange for photosynthates leaked from the host during photosynthesis (Wagner-Döbler et al., 2010). This finding is in line with those of previous studies showing that some species of microalgae require vitamins B1 and B12 as growth factors (Croft et al., 2006). In particular, Vitamin B12 has been identified as a growth promoter for microalgae, and exogenous addition of vitamin B12 has been shown to regulate microalgal growth by enhancing photosynthetic performance (Li et al., 2007; Helliwell et al., 2011; Jalilian et al., 2019). Moreover, some macroalgae species, such as Porphyra tenera and Bangia fuscopurpurea, require vitamin B12 during growth (Kendra and Hadwiger, 1984; Croft et al., 2005).
Proteobacteria have also been reported to deplete chitin and NAG (N-acetylglucosamine) (Cottrell and Kirchman, 2000b). Chitin deacetylation products, such as chitosan and chitooligosaccharide, have been shown to have antibacterial activity against plant pathogenic bacteria (Kendra and Hadwiger, 1984) and can induce plant resistance to pathogens in higher plant species (Pospieszny et al., 1991; Benhamou and Thériault, 1992; Struszczyk and Pospieszny, 1997). Additionally, members of the genera Zobellia, Cellulophaga, and Kordia within Proteobacteria have been found having algicidal activities, and have been suggested to control microalgal blooms (Sohn et al., 2004; Thomas et al., 2011). For example, an active substance called korkormicin, isolated from Pseudoalteromonas sp. F-420 inhibits bacterial growth by blocking the respiratory chain (Nakayama et al., 1999). Therefore, N. katadae may facilitate the establishment of dominant ecological niches for members of the phylum Proteobacteria in the epiphytic microbial community by providing favorable environmental conditions, thereby reducing the risk of pathogen infection.
4.2 Prediction functional profiles of bacterial communitiesSeveral highly abundant genes were observed in the functional prediction section, including K02014, which encodes an iron complex outer membrane receptor protein. This suggests that these microorganisms can enrich Fe for their own use and for the benefit of algae, which may promote algal growth. Previous studies have shown that Halomonas spp. can enhance the growth of the green alga Duniella balwardii under iron-deficient conditions, indicating that algae can utilize bacterial siderophores (Keshtacher-Liebso et al., 1995). Although epiphytic microorganisms can provide iron to the host, further experimental evidence is required to confirm their functionality.
Studies have demonstrated that magnetic field treatment during wheat seed germination can increase peroxidase isoenzyme and nitrate reductase activity in leaves (Marinković et al., 2008). Hydrogen peroxidase has a scavenging effect on free radicals and contains metal ions that are influenced by magnetic fields, leading to conformational changes and enzyme activation. Therefore, we hypothesized that microbial enrichment with metal ions may also enhance algal growth and promote peroxidase activity in algae, ultimately enhancing drought tolerance in N. yezoensis.
Several genes associated with the ABC transporter system were found to have higher bacterial abundances on the surface of N. yezoensis, including K06147 ABCB-BAC (ATP-binding cassette, sub-family E, member 1), K02003 ABC. CD. A (putative ABC transport system ATP-binding protein), K02004 ABC. CD. P (putative ABC transport system permease protein), K01990 ABC-2. A (ABC-2 type transport system ATP-binding protein), and K01992 ABC-2. P (ABC-2 type transport system permease protein). ABC transporters are membrane proteins that hydrolyze ATP to transport a wide range of substrates across the cellular membrane (Hollenstein et al., 2007). Bacteria play a role in nutrient uptake and toxin extrusion, contributing to drug and antibiotic resistance in microbial pathogens (Davidson and Chen, 2004). The presence of these ABC transporter genes on the surface of N. yezoensis suggests that they may provide metabolic energy and contribute to bacterial attachment, thereby establishing a bridge for material exchange between bacteria and N. yezoensis.
Other genes with high abundance on the surface of N. yezoensis were identified as K02342 dnaQ (DNA polymerase Ⅲ subunit epsilon), K03088 rpoE (RNA polymerase sigma-70 factor, ECF subfamily), and K06180 rluD (23S rRNA pseudouridine1911/1915/1917 synthase). This suggests that the surface flora of N. yezoensis exhibited a more active proliferation.
Three genes, K01091 gph (phosphoglycolate phosphatase), K03406 mcp (methyl-accepting chemotaxis protein), and K00626 ACAT (acetyl-CoA C-acetyltransferase) were more abundant in the surface bacteria of N. katadae. Gph encodes an enzyme belonging to the haloacid dehalogenase (HAD)-like phosphatase family, which has 2-phosphoglycolate phosphatase activity (Lyngstadaas et al., 1995). 2-Phosphoglycolate is generated during DNA strand break repair with 3′-phosphoglycolate ends, and such breaks can be induced by radiomimetic drugs, such as bleomycin. Gph has been implicated in the recovery of glycolate from 2-phosphoglycolate released by DNA repair enzymes following bleomycin treatment (Lyngstadaas et al., 1999; Pellicer et al., 2003; Kuznetsova et al., 2006). Methyl-accepting chemotaxis proteins (MCPs) such as K03406 mcp are transmembrane receptors that enable bacteria to sense various carbohydrates, including maltose, galactose, and ribose, as chemoattractants (Lux et al., 1995). It has been proposed that MCP-related genes may play a role in the recruitment of beneficial microorganisms to plant roots, leading to the suppression of soil-borne pathogens (Gao et al., 2021). Acetyl-CoA C-acetyltransferase, encoded by the K00626 ACAT gene, is involved in pathways such as fatty acid degradation; valine, leucine, and isoleucine degradation; and fatty acid metabolism, which are crucial for maintaining normal physiological functions in bacteria. Furthermore, a small number of transport and vitamin B12 genes were also detected, indicating that the associated microbiota may produce regulatory compounds and transport them to the algal host, as reported in previous studies, demonstrating that algae acquire vitamin B12 through symbiotic relationships with bacteria (Cruz-López and Maske, 2016). However, this gene was barely enriched in N. yezoensis.
PWY-3781 (aerobic respiration Ⅰ (cytochrome c)), PWY-7664 (oleate biosynthesis Ⅳ (anaerobic)), and PWY-5667 (CDP-diacylglycerol biosynthesis Ⅰ) were more abundant in N. katadae than in N. yezoensis. The main function of the respiratory chains in aerobically grown bacteria is to generate an electrochemical proton gradient across the cytoplasmic membrane through the translocation of protons, which is then utilized for ATP synthesis, solute uptake, and other energy-requiring membrane-associated processes, such as flagellar motion (Anraku and Gennis, 1987). Additionally, the respiratory chain regulates substrate-level metabolism through an energy-charge control mechanism, making it crucial for both material and energy metabolism in cells. Previous studies have shown a close relationship between the mitochondrial respiratory chain and the disease-resistance response in plants (Lennon et al., 1997; Lam et al., 2001).
Oleate is a monounsaturated fatty acid that plays a crucial role in the biosynthesis of unsaturated fatty acids (UFAs), which are essential for the structure and function of cell membranes, except in Archaea (Bi et al., 2013). Another pathway involved in membrane biosynthesis is the biosynthesis of CDP-diacylglycerol, which is found in eukaryotes and some gram-negative bacteria, particularly Gammaproteobacteria. CDP-diacylglycerol reacts with modifying compounds to form phosphoglycerides, which are important components of cell membranes (Bi et al., 2013). The relative abundance of DTDPRHAMSYN-PWY (dTDP-β-L-rhamnose biosynthesis) was found higher in N. yezoensis microorganisms than in N. katadae microorganisms. This pathway is widely conserved in Gram-positive and Gram-negative bacteria and is involved in the biosynthesis of L-rhamnopyranose, a deoxysugar that serves as a building block of the glycan component of lipopolysaccharides present in the enterobacterial common antigen (ECA) and O-antigens in many bacterial species, as well as a common component of the cell wall and capsule of pathogenic bacteria, which interact with the host during infection and are crucial for bacterial survival (Giraud and Naismith, 2000).
The three pathways enriched in N. katadae included those involved in bacterial energy metabolism and physiological structure formation, likely because of the absence of desiccation and the mutualistic relationship between epiphytic bacteria and algae. In contrast, the pathways in N. yezoensis indicate that bacteria are under drought stress due to desiccation, which may lead to bacterial death and separation from the algal surface upon re-entry into water. However, the presence of rhamnose in the cell walls and capsules of many pathogenic bacteria may contribute to their resistance to phagocytosis, drought, and adhesion, allowing them to remain attached to algae and regain their physiological activity after exposure to water (Giraud and Naismith, 2000). This observation is consistent with the higher proportion of surface epiphytic microorganisms belonging to potentially pathogenic bacterial phyla in N. yezoensis than in N. katadae. The surface microorganisms of the two algal species displayed clear differences in both structure and quantity. Furthermore, the genes and pathways enriched in each group exhibited significant differences. These variations can likely be attributed to factors such as the growth habits of the algae as well as their unique physical and chemical properties.
4.3 Bacterial community abundanceThe results of the real-time quantitative PCR (Fig. 9) revealed that the bacterial copy numbers on the surface of N. yezoensis were significantly higher than those on the surface of N. katadae, with a 28-fold difference at location 1 and a 4-fold difference at location 2. This indicates that N. yezoensis harbors a greater abundance of epiphytic bacteria than N. katadae, which may be attributed to the characteristics and physiological properties of the algae. N. yezoensis is thinner and may secrete more polysaccharides during growth, which can attract sediment particles and bacteria upon exposure to water. Furthermore, the desiccation phenomenon in N. yezoensis, in which gradual evaporation of the water film on the algal surface leads to cell damage and content outflow, could provide favorable conditions for bacterial attachment. Previous studies have also reported higher abundances of agar-degrading bacteria in diseased N. yezoensis compared to healthy N. yezoensis (Davison and Pearson, 1996; Manz et al., 1996; Cottrell and Kirchman, 2000b; Hollenstein et al., 2007). In contrast, N. katadae has a thicker body, a smoother surface, and does not experience desiccation during its life cycle. It can still grow in shallow puddles, even in high-tide zones, which limits bacterial exposure. Thus, there are fewer epiphytic bacteria on the surface of N. katadae than on N. yezoensis.
5 CONCLUSIONAlgae and bacteria in the ocean are inextricably linked and have intricate interactions that constitute important regulators of the structure and function of marine ecosystems. On the one hand, algae provide nutrients to bacteria, both dissolved organic matter (DOM) and particulate organic matter (POM) released by algae can be used by bacteria. However, bacteria create a good environment for algal growth and help to form or maintain algal forms. Our study demonstrated that N. yezoensis and N. katadae growing at the same tidal level in the same location differed greatly both in terms of species and the abundance of bacteria on their surfaces. By studying the species and abundance of bacterial communities, it can be concluded that specific metabolic pathways and enriched genes play important roles in interactions between algal and microbial communities. Future study shall focus on the effects of the physiological traits of algae on the microbial community, and the diverse effects of the functional characteristics of the microbial community on its hosts. It shall be emphasized that our results and conclusions are based on bioinformatic methods; further experimental evidence is required to confirm their functionality in this regard.
6 DATA AVAILABILITY STATEMENTData are available on request due to privacy or other restrictions.
Electronic supplementary material
Supplementary material (Supplementary Table S1 and Fig.S1) is available in the online version of this article at https://doi.org/10.1007/s00343-023-3078-5.
Ahmed A, Khurshid A, Tang X H, et al. 2021. Structural and functional impacts of microbiota on Pyropia yezoensis and surrounding seawater in cultivation farms along coastal areas of the Yellow Sea. Microorganisms, 9(6): 1291.
DOI:10.3390/microorganisms9061291 |
Alonso C, Warnecke F, Amann R, et al. 2007. High local and global diversity of Flavobacteria in marine plankton. Environmental Microbiology, 9(5): 1253-1266.
DOI:10.1111/j.1462-2920.2007.01244.x |
Anraku Y, Gennis R B. 1987. The aerobic respiratory chain of Escherichia coli. Trends in Biochemical Sciences, 12: 262-266.
DOI:10.1016/0968-0004(87)90131-9 |
Bathnagar D, Deb A R. 1978. Some effects of pregermination exposure of wheat seeds to magnetic field. Ⅱ. Effect on some physiological processes. Seed Research, 6(1): 14-22.
|
Benhamou N, Thériault G. 1992. Treatment with chitosan enhances resistance of tomato plants to the crown and root rot pathogen Fusarium oxysporum f. sp. radicis-lycopersici. Physiological and Molecular Plant Pathology, 41(1): 33-52.
DOI:10.1016/0885-5765(92)90047-Y |
Bernardet J F, Segers P, Vancanneyt M, et al. 1996. Cutting a Gordian knot: emended classification and description of the genus Flavobacterium, emended description of the family Flavobacteriaceae, and proposal of Flavobacterium hydatis nom. nov. (Basonym, Cytophaga aquatilis Strohl and Tait 1978). International Journal of Systematic Bacteriology, 46(1): 128-148.
DOI:10.1099/00207713-46-1-128 |
Bi H K, Wang H H, Cronan J E. 2013. FabQ, a dual-function dehydratase/isomerase, circumvents the last step of the classical fatty acid synthesis cycle. Chemistry & Biology, 20(9): 1157-1167.
DOI:10.1016/j.chembiol.2013.07.007 |
Biegala I C, Kennaway G, Alverca E, et al. 2002. Identification of bacteria associated with Dinoflagellates (Dinophyceae) Alexandrium spp. using tyramide signal amplification-fluorescent in situ hybridization and confocal microscopy. Journal of Phycology, 38(2): 404-411.
DOI:10.1046/j.1529-8817.2002.01045.x |
Bolyen E, Rideout J R, Dillon M R, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37(8): 852-857.
DOI:10.1038/s41587-019-0209-9 |
Bulleri F, Chapman M G. 2010. The introduction of coastal infrastructure as a driver of change in marine environments. Journal of Applied Ecology, 47(1): 26-35.
DOI:10.1111/j.1365-2664.2009.01751.x |
Callahan B J, McMurdie P J, Rosen M J, et al. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nature Methods, 13(7): 581-583.
DOI:10.1038/nmeth.3869 |
Campbell A H, Marzinelli E M, Gelber J, et al. 2015. Spatial variability of microbial assemblages associated with a dominant habitat-forming seaweed. Frontiers in Microbiology, 6: 230.
DOI:10.3389/fmicb.2015.00230 |
Chapman M G, Underwood A J. 1999. Ecological patterns in multivariate assemblages: information and interpretation of negative values in ANOSIM tests. Marine Ecology Progress Series, 180: 257-265.
DOI:10.3354/meps180257 |
Chen Z J, Shao Y, Li Y J, et al. 2020. Rhizosphere bacterial community structure and predicted functional analysis in the water-level fluctuation zone of the Danjiangkou Reservoir in China during the dry period. International Journal of Environmental Research and Public Health, 17(4): 1266.
DOI:10.3390/ijerph17041266 |
Cook K, Vanderklift M A, Poore A G B. 2011. Strong effects of herbivorous amphipods on epiphyte biomass in a temperate seagrass meadow. Marine Ecology Progress Series, 442: 263-269.
DOI:10.3354/meps09446 |
Cosgrove D J. 2005. Growth of the plant cell wall. Nature Reviews Molecular Cell Biology, 6(11): 850-861.
DOI:10.1038/nrm1746 |
Cottrell M T, Kirchman D L. 2000a. Community composition of marine bacterioplankton determined by 16S rRNA gene clone libraries and fluorescence in situ hybridization. Applied and Environmental Microbiology, 66(12): 5116-5122.
DOI:10.1128/AEM.66.12.5116-5122.2000 |
Cottrell M T, Kirchman D L. 2000b. Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Applied and Environmental Microbiology, 66(4): 1692-1697.
DOI:10.1128/AEM.66.4.1692-1697.2000 |
Croft M T, Lawrence A D, Raux-Deery E, et al. 2005. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature, 438(7064): 90-93.
DOI:10.1038/nature04056 |
Croft M T, Warren M J, Smith A G. 2006. Algae need their vitamins. Eukaryotic Cell, 5(8): 1175-1183.
DOI:10.1128/EC.00097-06 |
Cruz-López R, Maske H. 2016. The vitamin B1 and B12 required by the marine dinoflagellate Lingulodinium polyedrum can be provided by its associated bacterial community in culture. Frontiers in Microbiology, 7: 560.
DOI:10.3389/fmicb.2016.00560 |
Davidson A L, Chen J. 2004. ATP-binding cassette transporters in bacteria. Annual Review of Biochemistry, 73(1): 241.
DOI:10.1146/annurev.biochem.73.011303.073626 |
Davison I R, Pearson G A. 1996. Stress tolerance in intertidal seaweeds. Journal of Phycology, 32(2): 197-211.
DOI:10.1111/j.0022-3646.1996.00197.x |
Delattre C, Fenoradosoa T A, Michaud P. 2011. Galactans: an overview of their most important sourcing and applications as natural polysaccharides. Brazilian Archives of Biology and Technology, 54(6): 1075-1092.
DOI:10.1590/S1516-89132011000600002 |
Douglas G M, Maffei V J, Zaneveld J, et al. 2019. PICRUSt2: an improved and extensible approach for metagenome inference. BioRxiv.
DOI:10.1101/672295 |
Egan S, Thomas T. 2015. Editorial for: microbial symbiosis of marine sessile hosts-diversity and function. Frontiers in Microbiology, 6: 585.
DOI:10.3389/fmicb.2015.00585 |
Falkowski P G, Katz M E, Knoll A H, et al. 2004. The evolution of modern eukaryotic phytoplankton. Science, 305(5682): 354-360.
DOI:10.1126/science.1095964 |
Gao M, Xiong C, Gao C, et al. 2021. Disease-induced changes in plant microbiome assembly and functional adaptation. Microbiome, 9(1): 187.
DOI:10.1186/s40168-021-01138-2 |
Giraud M F, Naismith J H. 2000. The rhamnose pathway. Current Opinion in Structural Biology, 10(6): 687-696.
DOI:10.1016/S0959-440X(00)00145-7 |
Goecke F, Labes A, Wiese J, et al. 2010. Chemical interactions between marine macroalgae and bacteria. Marine Ecology Progress Series, 409: 267-299.
DOI:10.3354/meps08607 |
Haas B J, Gevers D, Earl A M, et al. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research, 21(3): 494-504.
DOI:10.1101/gr.112730.110 |
Hartman W H, Ye R Z, Horwath W R, et al. 2017. A genomic perspective on stoichiometric regulation of soil carbon cycling. The ISME Journal, 11(12): 2652-2665.
DOI:10.1038/ismej.2017.115 |
Helliwell K E, Wheeler G L, Leptos K C, et al. 2011. Insights into the evolution of vitamin B12 auxotrophy from sequenced algal genomes. Molecular Biology and Evolution, 28(10): 2921-2933.
DOI:10.1093/molbev/msr124 |
Hollenstein K, Dawson R J, Locher K P. 2007. Structure and mechanism of ABC transporter proteins. Current Opinion in Structural Biology, 17(4): 412-418.
DOI:10.1016/j.sbi.2007.07.003 |
Jalilian N, Najafpour G D, Khajouei M. 2019. Enhanced vitamin B12 production using Chlorella vulgaris. International Journal of Engineering, 32(1): 1-9.
DOI:10.5829/ije.2019.32.01a.01 |
Ji Y, Xu Z G, Zou D H, et al. 2016. Ecophysiological responses of marine macroalgae to climate change factors. Journal of Applied Phycology, 28(5): 2953-2967.
DOI:10.1007/s10811-016-0840-5 |
Johansen J E, Nielsen P, Sjøholm C. 1999. Description of Cellulophaga baltica gen. nov., sp. nov. and Cellulophaga fucicola gen. nov., sp. nov. and reclassification of [Cytophaga] lytica to Cellulophaga lytica gen. nov., comb. nov. International Journal of Systematic and Evolutionary Microbiology, 49(3): 1231-1240.
DOI:10.1099/00207713-49-3-1231 |
Kendra D F, Hadwiger L A. 1984. Characterization of the smallest chitosan oligomer that is maximally antifungal to Fusarium solani and elicits pisatin formation in Pisum sativum. Experimental Mycology, 8(3): 276-281.
DOI:10.1016/0147-5975(84)90013-6 |
Keshtacher-Liebso E, Hadar Y, Chen Y. 1995. Oligotrophic bacteria enhance algal growth under iron-deficient conditions. Applied and Environmental Microbiology, 61(6): 2439-2441.
DOI:10.1128/aem.61.6.2439-2441.1995 |
Kuznetsova E, Proudfoot M, Gonzalez C F, et al. 2006. Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. Journal of Biological Chemistry, 281(47): 36149-36161.
DOI:10.1074/jbc.M605449200 |
Lam E, Kato N, Lawton M. 2001. Programmed cell death, mitochondria and the plant hypersensitive response. Nature, 411(6839): 848-853.
DOI:10.1038/35081184 |
Langille M G I, Zaneveld J, Caporaso J G, et al. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 31(9): 814-821.
DOI:10.1038/nbt.267 |
Lebrun E S, Kang S. 2018. A comparison of computationally predicted functional metagenomes and microarray analysis for microbial P cycle genes in a unique basalt-soil forest. F1000Research, 7: 179.
DOI:10.12688/f1000research.13841.1 |
Lennon A M, Neuenschwander U H, Ribas-Carbo M, et al. 1997. The effects of salicylic acid and tobacco mosaic virus infection on the alternative oxidase of tobacco. Plant Physiology, 115(2): 783-791.
DOI:10.1104/pp.115.2.783 |
Li B, Zhang X X, Guo F, et al. 2013. Characterization of tetracycline resistant bacterial community in saline activated sludge using batch stress incubation with high-throughput sequencing analysis. Water Research, 47(13): 4207-4216.
DOI:10.1016/j.watres.2013.04.021 |
Li T D, Doronina N V, Ivanova E G, et al. 2007. Vitamin B12-independent strains of Methylophaga marina isolated from Red Sea algae. Microbiology, 76(1): 75-81.
DOI:10.1134/S0026261707010110 |
Lin A P, Wang G C, Yang F, et al. 2009. Photosynthetic parameters of sexually different parts of Porphyra katadai var. hemiphylla (Bangiales, Rhodophyta) during dehydration and re-hydration. Planta, 229(4): 803-810.
DOI:10.1007/s00425-008-0874-2 |
Lux R, Jahreis K, Bettenbrock K, et al. 1995. Coupling the phosphotransferase system and the methyl-accepting chemotaxis protein-dependent chemotaxis signaling pathways of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America, 92(25): 11583-11587.
DOI:10.1073/pnas.92.25.11583 |
Lyngstadaas A, Løbner-Olesen A, Boye E. 1995. Characterization of three genes in the dam-containing operon of Escherichia coli. Molecular and General Genetics MGG, 247(5): 546-554.
DOI:10.1007/BF00290345 |
Lyngstadaas A, Løbner-Olesen A, Grelland E, et al. 1999. The gene for 2-phosphoglycolate phosphatase (gph) in Escherichia coli is located in the same operon as dam and at least five other diverse genes. Biochimica et Biophysica Acta (BBA) -General Subjects, 1472(1-2): 376-384.
DOI:10.1016/S0304-4165(99)00146-4 |
Magoč T, Salzberg S L. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27(21): 2957-2963.
DOI:10.1093/bioinformatics/btr507 |
Manz W, Amann R, Ludwig W, et al. 1996. Application of a suite of 16S rRNA-specific oligonucleotide probes designed to investigate bacteria of the phylum cytophaga-flavobacter-bacteroides in the natural environment. Microbiology, 142(5): 1097-1106.
DOI:10.1099/13500872-142-5-1097 |
Marinković B, Grujić M, Marinković D, et al. 2008. Use of biophysical methods to improve yields and quality of agricultural products. Journal of Agricultural Sciences, Belgrade, 53(3): 235-242.
DOI:10.2298/JAS0803235M |
Mercado J, Niell F X. 2000. Carbon dioxide uptake by Bostrychia scorpioides (Rhodophyceae) under emersed conditions. British Phycological Bulletin, 35(1): 45-51.
DOI:10.1080/09670260010001735611 |
Nakayama Y, Hayashi M, Yoshikawa K, et al. 1999. Inhibitor studies of a new antibiotic, korormicin, 2-n-heptyl-4-hydroxyquinoline N-oxide and Ag+ toward the Na+-translocating NADH-quinone reductase from the marine Vibrio alginolyticus. Biological and Pharmaceutical Bulletin, 22(10): 1064-1067.
DOI:10.1248/bpb.22.1064 |
Nichols C M, Bowman J P, Guezennec J. 2005. Olleya marilimosa gen. nov., sp. nov., an exopolysaccharide-producing marine bacterium from the family Flavobacteriaceae, isolated from the Southern Ocean. International Journal of Systematic and Evolutionary Microbiology, 55(4): 1557-1561.
DOI:10.1099/ijs.0.63642-0 |
Pellicer M T, Nuñez M F, Aguilar J, et al. 2003. Role of 2-phosphoglycolate phosphatase of Escherichia coli in metabolism of the 2-phosphoglycolate formed in DNA repair. Journal of Bacteriology, 185(19): 5815-5821.
DOI:10.1128/JB.185.19.5815-5821.2003 |
Pospieszny H, Chirkov S, Atabekov J. 1991. Induction of antiviral resistance in plants by chitosan. Plant Science, 79(1): 63-68.
DOI:10.1016/0168-9452(91)90070-O |
Provasoli L, Carlucci A. 1974. Vitamins and growth regulators. In: Stewart W D P ed. Algal Physiology and Biochemistry, Botanical Monographs. University of California Press, Berkeley. p. 741-787.
|
Ribeiro H, de Sousa T, Santos J P, et al. 2018. Potential of dissimilatory nitrate reduction pathways in polycyclic aromatic hydrocarbon degradation. Chemosphere, 199: 54-67.
DOI:10.1016/j.chemosphere.2018.01.171 |
Rusch D B, Halpern A L, Sutton G, et al. 2007. The Sorcerer II global ocean sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biology, 5(3): e77.
DOI:10.1371/journal.pbio.0050077 |
Schäfer H, Abbas B, Witte H, et al. 2002. Genetic diversity of 'satellite' bacteria present in cultures of marine diatoms. FEMS Microbiology Ecology, 42(1): 25-35.
DOI:10.1111/j.1574-6941.2002.tb00992.x |
Segata N, Izard J, Waldron L, et al. 2011. Metagenomic biomarker discovery and explanation. Genome Biology, 12(6): R60.
DOI:10.1186/gb-2011-12-6-r60 |
Simonato F, Gómez-Pereira P R, Fuchs B M, et al. 2010. Bacterioplankton diversity and community composition in the Southern Lagoon of Venice. Systematic and Applied Microbiology, 33(3): 128-138.
DOI:10.1016/j.syapm.2009.12.006 |
Sohn J H, Lee J H, Yi H N, et al. 2004. Kordia algicida gen. nov., sp. nov., an algicidal bacterium isolated from red tide. International Journal of Systematic and Evolutionary Microbiology, 54(3): 675-680.
DOI:10.1099/ijs.0.02689-0 |
Stat M, Pochon X, Franklin E C, et al. 2013. The distribution of the thermally tolerant symbiont lineage (Symbiodinium clade D) in corals from Hawaii: correlations with host and the history of ocean thermal stress. Ecology and Evolution, 3(5): 1317-1329.
DOI:10.1002/ece3.556 |
Struszczyk H, Pospieszny H. 1997. New Applications of Chitinand Its Derivatives in Plant Protection. CRC Press. Boca Raton, Florida, 14p.
|
Thomas F, Hehemann J H, Rebuffet E, et al. 2011. Environmental and gut Bacteroidetes: the food connection. Frontiers in Microbiology, 2: 93.
DOI:10.3389/fmicb.2011.00093 |
Tujula N A, Crocetti G R, Burke C, et al. 2010. Variability and abundance of the epiphytic bacterial community associated with a green marine Ulvacean alga. The ISME Journal, 4(2): 301-311.
DOI:10.1038/ismej.2009.107 |
Venter J C, Remington K, Heidelberg J F, et al. 2004. Environmental genome shotgun sequencing of the Sargasso Sea. Science, 304(5667): 66-74.
DOI:10.1126/science.1093857 |
Wagner M R, Lundberg D S, Coleman-Derr D, et al. 2014. Natural soil microbes alter flowering phenology and the intensity of selection on flowering time in a wild Arabidopsis relative. Ecology Letters, 17(6): 717-726.
DOI:10.1111/ele.12276 |
Wagner-Döbler I, Ballhausen B, Berger M, et al. 2010. The complete genome sequence of the algal symbiont Dinoroseobacter shibae: a hitchhiker's guide to life in the sea. The ISME Journal, 4(1): 61-77.
DOI:10.1038/ismej.2009.94 |
Wang W J, Sun X T, Liu F L, et al. 2016. Effect of abiotic stress on the gameophyte of Pyropia katadae var. hemiphylla (Bangiales, Rhodophyta). Journal of Applied Phycology, 28(1): 469-479.
DOI:10.1007/s10811-015-0579-4 |
Yoshida T, Notoya M, Kikuchi N, et al. 1997. Catalogue of species of Porphyra in the world, with special reference to the type locality and bibliography. Natural History Research., 3: 5-18.
|